Professor Ravi Radhakrishnan has a new paper out in the Proceedings of the National Academy of Sciences of the United States of America.
Significance
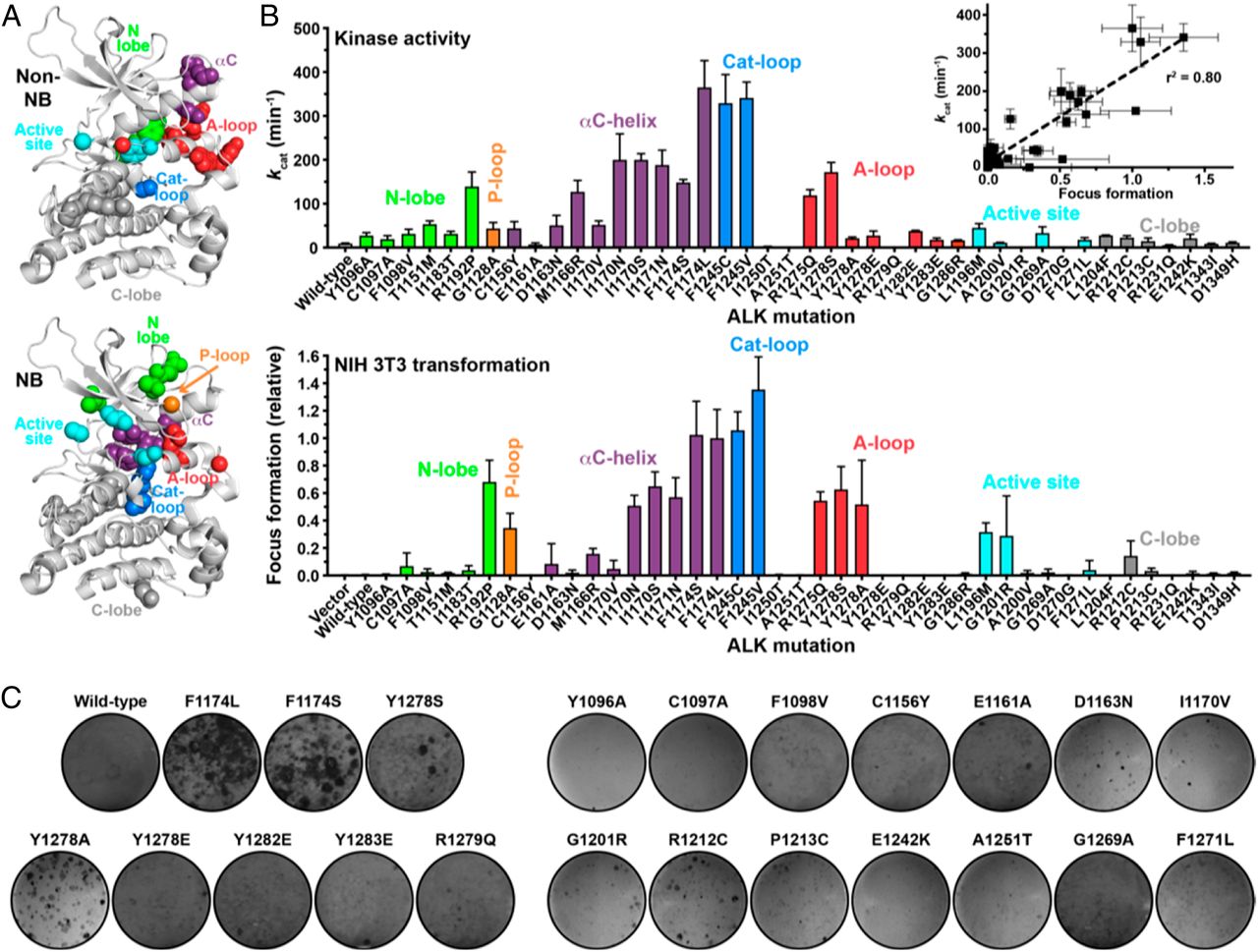
High-risk tumors are genomically heterogeneous, harboring gene amplifications and mutations. The activation status of mutated proteins in cancer can profoundly impact disease progression, patient response, and drug sensitivity. Yet, outside of a few hotspot mutations, functional studies of clinically observed mutations are not commonly pursued. We report a combined experimental profiling and computational analysis of the effects of clinically observed and “test” mutations in the kinase domain of anaplastic lymphoma kinase (ALK), a known oncogenic driver in pediatric neuroblastoma. We find that the activation status of the mutated protein is a good indicator of the transforming ability in NIH 3T3 cells. We also report biophysical as well as data-driven models with predictive power to profile these mutant kinases in silico.
Abstract
Kinases play important roles in diverse cellular processes, including signaling, differentiation, proliferation, and metabolism. They are frequently mutated in cancer and are the targets of a large number of specific inhibitors. Surveys of cancer genome atlases reveal that kinase domains, which consist of 300 amino acids, can harbor numerous (150 to 200) single-point mutations across different patients in the same disease. This preponderance of mutations—some activating, some silent—in a known target protein make clinical decisions for enrolling patients in drug trials challenging since the relevance of the target and its drug sensitivity often depend on the mutational status in a given patient. We show through computational studies using molecular dynamics (MD) as well as enhanced sampling simulations that the experimentally determined activation status of a mutated kinase can be predicted effectively by identifying a hydrogen bonding fingerprint in the activation loop and the αC-helix regions, despite the fact that mutations in cancer patients occur throughout the kinase domain. In our study, we find that the predictive power of MD is superior to a purely data-driven machine learning model involving biochemical features that we implemented, even though MD utilized far fewer features (in fact, just one) in an unsupervised setting. Moreover, the MD results provide key insights into convergent mechanisms of activation, primarily involving differential stabilization of a hydrogen bond network that engages residues of the activation loop and αC-helix in the active-like conformation (in >70% of the mutations studied, regardless of the location of the mutation).